Search

Wal-yan Respiratory Research Centre PhD student Niamh Troy has found how OM85 helps babies fight off severe lung infections.

Nearly 50 existing prescription medications already used by Australians will be tested by new research in the fight against COVID’s mutant variants.
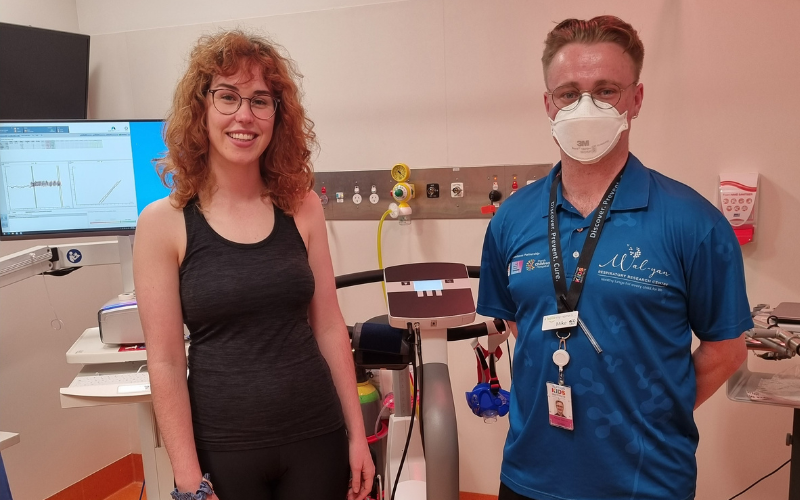
Natasha, who is a participant in the West Australian Lung Health in Prematurity (WALHIP) study, this week became the first person to receive a lung health assessment using new state-of-the-art lung function testing equipment at Perth Children’s Hospital.

A project to be undertaken by a team of researchers at the Wal-yan Respiratory Research Centre, led by chief investigator Professor Stephen Stick, aims to develop interventions that could provide protection in the event of a new pandemic, and against common viruses already infecting children in WA.

Wal-yan Respiratory Research Centre team members and special guests travelled to Wadjemup (Rottnest) on 27 and 28 October to spend an intensive two days together learning about, and providing input into, the broad range of research projects underway within the Centre.

A research program, which enables over 25 important respiratory research studies to be undertaken, celebrated the recruitment of its 300th participant on 14 December 2022.

Seven innovative lung health research projects have received funding support as the 2023 Wal-yan Respiratory Research Centre Strategic Inspiration Projects.

More than 14 researchers from the Wal-yan Respiratory Research Centre will be welcomed as presenters and facilitators at The Thoracic Society of Australia and New Zealand and The Australia and New Zealand Society of Respiratory Science (TSANZSRS) Annual Scientific Meeting (ASM) this weekend.

Patients with lung infections that are not responding to antibiotics will be treated with phage therapy as part of a translational trial program to be undertaken by world-recognised experts in this field.

The Wal-yan Respiratory Research Centre this month welcomed new PhD scholarship awardee Yaqin Alziyadat, whose exciting research work will support the Centre’s vision to ensure all children have healthy lungs for life.